

 联系我们
联系我们
铁死亡与神经退行性疾病、肿瘤、动脉硬化、糖尿病、急性肾损伤等多种疾病相关,致病机理复杂。通过适当的手段,激活或抑制铁死亡,可干预疾病的发展。因此铁死亡成为近年来的研究热点。本期内容将围绕这一热点,以文献为例展开讲述,分享一套关于铁死亡常用的研究思路和方法,并总结了关于铁死亡分子机制的几个关键途径。
一、研究思路和方法
1. 研究思路
目前铁死亡的研究方法基本遵循:
表型→蛋白表达→分子表达→基因敲除/过表达逆转表型
2. 研究方法
1. 形态特征检测
2. 基因表达检测
3. 蛋白水平检测
4. 生化特征指标检测
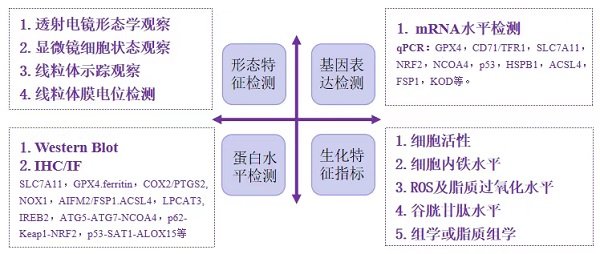
图1.铁死亡研究方法总结
3. 分子机制
1. GPX4与脂质过氧化
2. 胱氨酸/谷氨酸转运蛋白(System xc-)
3. 铁代谢作用
4. p53调控作用
5. VDACs调控作用
6. 其他调节通路
二、铁死亡分子机制的关键途径
1. GPX4与脂质过氧化
GPX4是细胞内重要的抗氧化酶,能够降解脂质过氧化物,保护细胞免受氧化损伤。当GPX4活性被抑制时,脂质过氧化物大量累积,诱导铁死亡。
2. 胱氨酸/谷氨酸转运蛋白(System xc-)
System xc-是细胞内一个重要的抗氧化系统它通过转运胱氨酸和谷氨酸来维持 GSH的合成当Svstem xc-功能受损时,胱氨酸摄取减少GSH合成受阻,GPX4活性下降,进而促进铁死亡。
3. 铁代谢作用
铁离子的积累是铁死亡发生的关键因素之一。铁离子通过催化脂质过氧化反应,增加ROS的生成,从而加速细胞死亡。细胞内铁的来源包括血红素加氧酶1(HO-1)和转铁蛋白等,它们通过调控铁代谢参与铁死亡的调节。
4. p53调控作用
p53作为一种抑癌基因,通过下调System xc-组分SLC7A11的表达,抑制细胞对胱氨酸的摄取,进而降低GSH和GPX4的水平,增强细胞对铁死亡的敏感性。
5. VDACs调控作用
VDACs是转运离子和代谢产物的跨膜通道。Erastin(铁死亡诱导剂)的铁死亡诱导机制与ROS和铁依赖性信号传导有关,Erastin 能够抑制VDAC2和VDAC3,加速氧化,导致内源活性氧积累。Erastin还破坏线粒体通透性过渡孔 (mPTP),引起线粒体功能紊乱,氧化性物质释放,最终引起细胞氧化性死亡。
6. 其他调节通路
在氧化应激状态下,甲硫氨酸可通过硫转移途径转化为胱氨酸,合成谷胱甘肽,协助谷胱甘肽过氧化物酶抑制脂质活性氧生成,避免氧化性细胞损伤.因此硫转移途径可抑制铁死亡的发生;血红素加氧酶是细胞内铁的重要来源之一,已被证实可以诱导脂质过氧化反应从而导致铁死亡的发生;转铁蛋白也是细胞内铁的来源之一,它亦参与了铁死亡的调节过程。
三、什么是铁死亡?
1.铁死亡机制
铁死亡(Ferroptosis)是一种独特的程序性细胞死亡方式,在细胞形态,代谢和分子生物学方面,均有别于其他已知的细胞死亡方式。
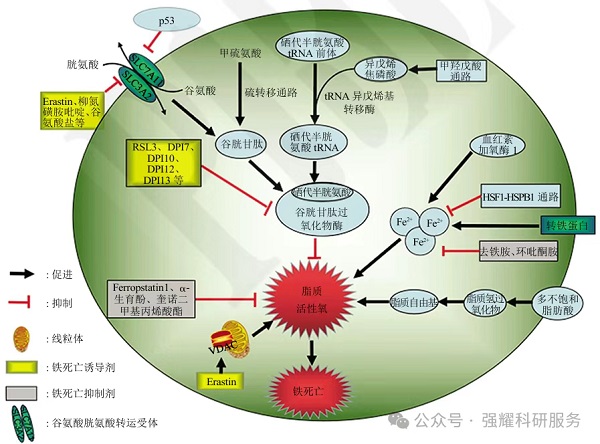
图2. 铁死亡发生机制
其发生机制如图2:其上游通路是通过直接或间接影响谷胱甘肽过氧化物酶(GPXs)的活性,降低细胞抗氧化能力,致使脂质过氧化反应增加,脂质活性氧增多,引起铁死亡的发生。此外,在Fe2+或酯氧合酶的作用下,催化细胞膜上高表达的不饱和脂肪酸,发生脂质过氧化,从而诱导细胞死亡。铁死亡的本质是谷胱甘肽的耗竭,谷胱甘肽过氧化物酶(GPX4)活性下降,脂质氧化物不能通过GPX4催化的谷胱甘肽还原酶反应代谢,之后Fe2+氧化脂质产生活性氧,从而促使铁死亡的发生。
2.主要特征
(1)细胞形态:铁死亡会导致细胞线粒体萎缩变小,嵴减少甚至消失,膜密度增高,细胞膜断裂和出泡,细胞核形态变化不明显。
(2)细胞成分:铁死亡表现为脂质过氧化增高,活性氧(ROS)升高,铁离子聚集,也有一些特征基因发生变化。
四、文献案例
铁死亡研究如此热门,在课题设计过程中,会涉及哪些方向的实验呢?我们以2021年刊登在Clinical and translational medicine杂志题为“Deubiquitinase USP35 modulates ferroptosis in lung cancer via targeting ferroportin”论文为例。
题目四要素:疾病(lung cancer)、表型(ferroptosis)、主变量分子(USP35)、机制(ferroportin)四部分内容。
研究思路及方法如下:
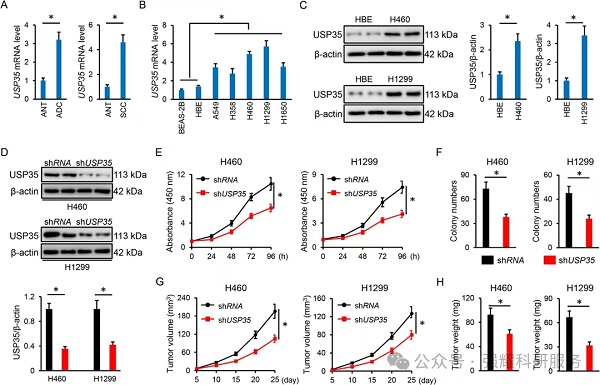
(1)现象
USP35基因的敲低可抑制肺癌细胞的生长、集落的形成和肿瘤的进展。
实验:CCK8检测,细胞克隆形成,肿瘤变化检测,WB检测,qPCR检测。
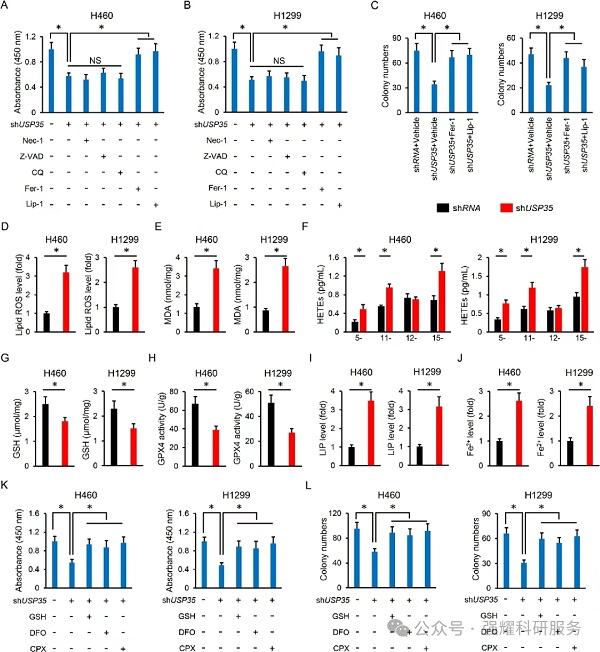
(2)本质
当干扰USP35的基因表达后,细胞活力下降,通过各种细胞死亡抑制剂处理后,发现细胞坏死,凋亡以及自噬抑制剂对细胞活力下降没有影响,但是铁死亡抑制剂能够缓解细胞死亡现象,初步考虑USP35影响细胞的铁死亡现象,然后通过铁死亡相关指标检测发现USP35敲低后,细胞铁死亡指标发生明显变化。
实验:CCK8检测,细胞克隆形成,脂质活性氧检测,HETEs检测,GSH以及GPX4检测,LIP检测。
Nec-1:细胞坏死抑制剂
Z-VAD:细胞凋亡抑制剂
CQ:自噬抑制剂
Fer-1:铁死亡抑制剂
Lip-1:铁死亡抑制剂DFO和CPX是铁螯合剂
DFO和CPX是铁螯合剂
(3)验证
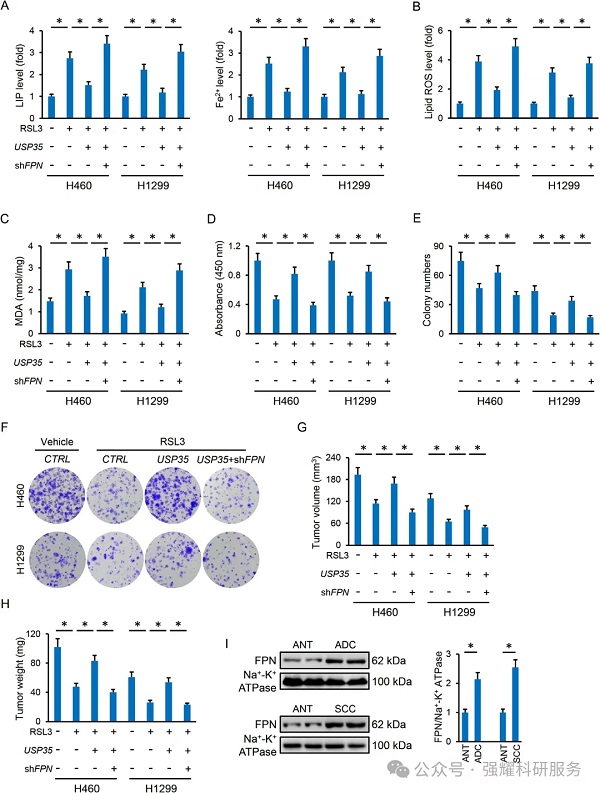
验证一:过表达USP35对癌细胞以及体内肿瘤没有明显作用;当处理铁死亡诱导剂的同时过表达USP35能发现不论是细胞还是在体肿瘤都进一步发展。USP35过表达阻断了erastin/rsl3介导的肿瘤抑制作用。
实验:CCK8检测,qPCR检测,细胞克隆形成,体内肿瘤实验
Erastin和RSL3铁死亡诱导剂
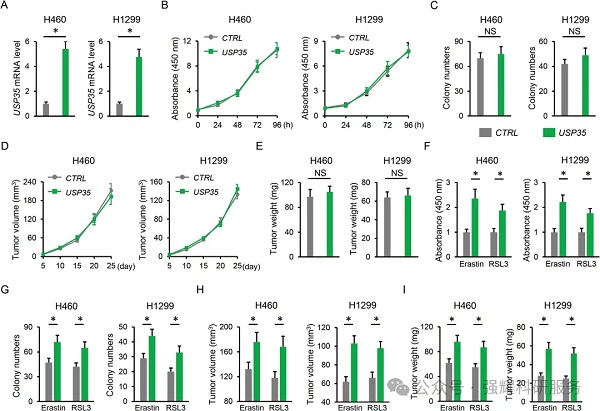
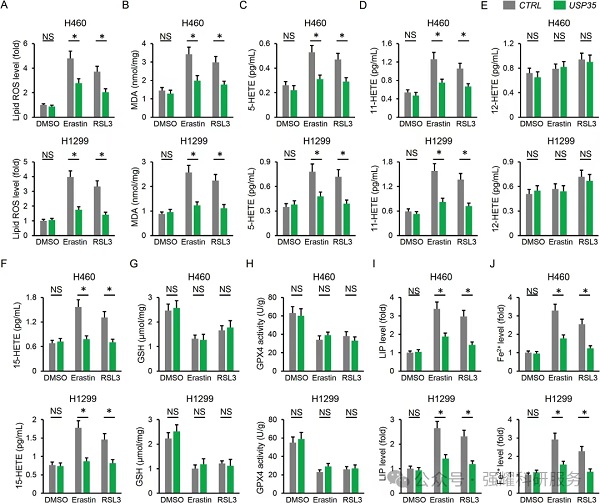
验证二:USP35的过表达能缓解Erastin和RSL3引起的铁死亡;能够降低Erastin和RSL3引起的铁蓄积,但对GSH和GPX4没有明显影响。
实验:脂质活性氧检测,HETEs检测,GSH以及GPX4检测,LIP检测,Fe2+检测。
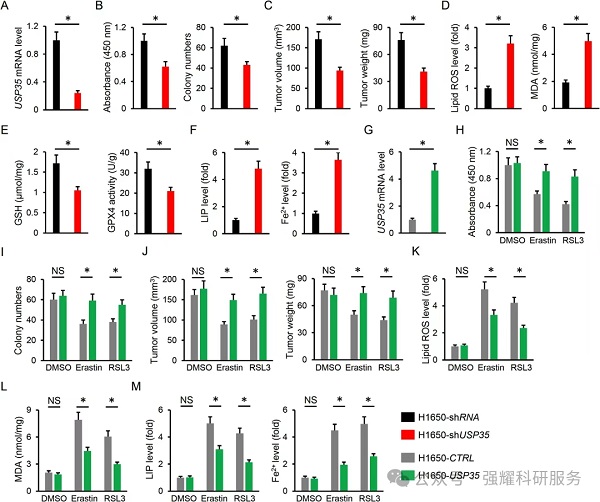
验证三:在H1650细胞中干扰USP35能够引起细胞活性减低和肿瘤生长抑制,并通过铁死亡指标发现是铁死亡现象;用Erastin和RSL3处理引起癌细胞铁死亡的同时过表达USP35能够促进细胞增殖和肿瘤发展,改善铁死亡现象。
实验:qPCR检测,CCK8检测,在体肿瘤检测,脂质活性氧检测,GSH以及GPX4检测,LIP检测,Fe2+检测。
(4)机制
机制一:USP35不影响GSH代谢相关蛋白也不影响cystine摄取,但是影响铁转运蛋白FPN(ferroportin);过表达USP35降低了肺癌细胞内的LIP和Fe2+,而在FPN缺陷的细胞中却没有降低。此外,在敲除内源性FPN后,过表达USP35所降低的脂质ROS和MDA的生成也被影响。
实验:WB检测,LIP检测,Fe2+检测。
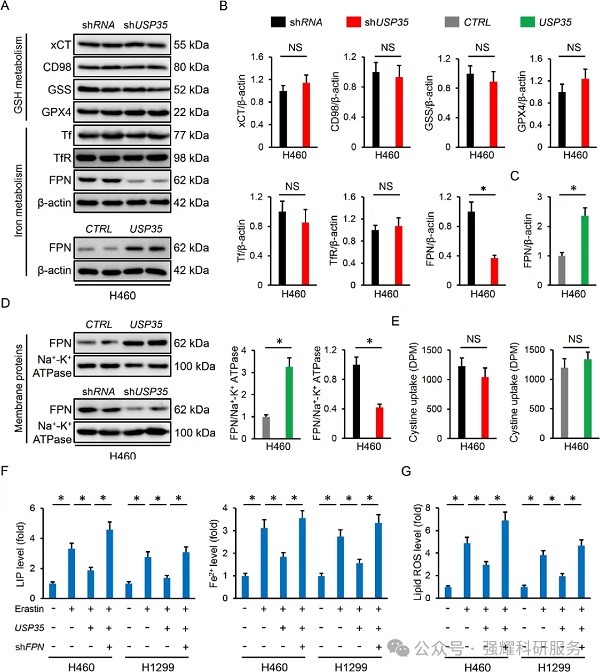
机制二:干扰FPN后,能够增加LIP和Fe2+的水平,增加ROS和MDA含量;通过CCK8,细胞克隆形成实验以及在体肿瘤检测,发现干扰FPN表达能够促进细胞铁死亡,抑制肿瘤进展;同时在肺腺癌以及肺鳞癌组织中FPN是高表达的。FPN是USP35调节铁死亡和肿瘤进展的潜在靶点。
实验:WB检测,CCK8检测,细胞克隆形成检测,在体肿瘤检测,LIP检测,Fe2+检测,脂质ROS检测,MDA检测。
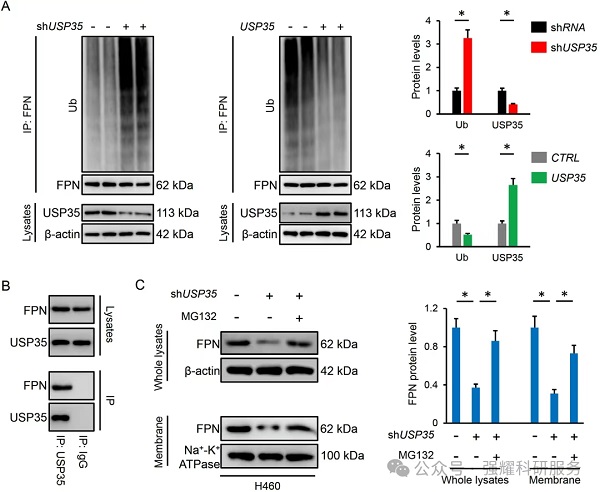
机制三:USP35敲低会增加,而过表达USP35降低了泛素化的FPN水平;IP实验证明FPN与内源性USP35能够结合,且通过MG132蛋白酶抑制剂证明了USP35可以直接与FPN相互作用,并作为一种去泛素酶来维持其蛋白的稳定性。
实验:WB检测,IP检测。
综上,到目前为止,仍然没有判定铁死亡的“金指标”,即决定性的代谢指标来判定一个细胞死亡模型是否为铁死亡,首先需要检测相关的指标(如亚铁、MDA、GSH/GSSG、GPX4、SLC7A11蛋白、PCG2等)是否出现变化,再使用铁死亡的抑制剂实验,观察是否能逆转细胞死亡和相关指标的变化。
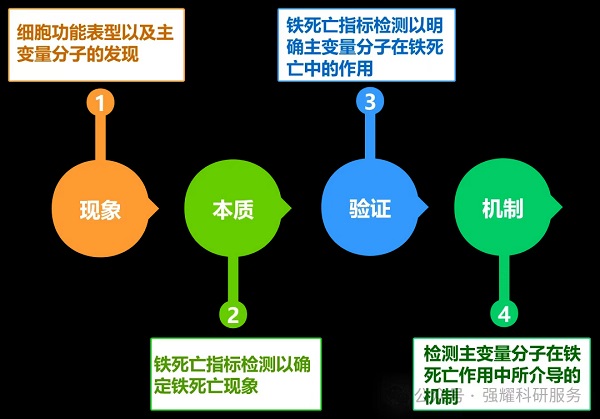
图3. 铁死亡研究思路借鉴
参考文献1:DOI:10.1016/j.cell.2022.06.003
参考文献2:DOI:10.1002/ctm2.390
强耀生物可提供蛋白、抗体制备、基因合成及测序等优秀服务助力科研
 返回
返回